Un equipo de investigadores del Centro de Huntington del University College de Londres ha desarrollado un fármaco que detiene el avance de la enfermedad de Huntington, un mal hereditario que desgasta algunas células nerviosas del cerebro, provoca graves problemas locomotores, cognitivos y emocionales, y reduce la esperanza de vida.
El medicamento se encuentra en la primera fase de pruebas, pero los primeros ensayos con enfermos han sido un éxito, según los científicos responsables del trabajo. El nuevo compuesto, al que sus desarrolladores han llamado IONIS-HTTRx, es el primer fármaco que logra disminuir la presencia en el cerebro de la proteína tóxica que desencadena esta enfermedad que en España padecen más de 4.000 personas.
46 razones para la esperanza
El medicamento se ha probado en 46 individuos de Alemania, Canadá y el Reino Unido que padecen la enfermedad de Huntington en su etapa inicial. A cada paciente se le inyectaron en el líquido cefalorraquídeo varias dosis de IONIS-HTTRx para que llegaran al cerebro, donde el fármaco redujo de forma considerable los niveles de huntingtina, la proteína que, al mutar, causa esta rara dolencia. El compuesto actúa sobre el gen que expresa la huntingtina, lo que inhibe la producción de esta, necesaria para muchas funciones, pero que al transformarse de forma maligna deteriora las células nerviosas.
Según Sarah Tabrizi, directora del Centro de Huntington del University College, “los resultados del ensayo son de vital importancia para los pacientes y sus familias. Es la primera vez que un fármaco reduce los niveles de la proteína tóxica que causa la dolencia, y el medicamento ha resultado seguro y tolerable para los enfermos. Ahora hay que hacer pruebas clínicas con más personas para confirmar con seguridad que el compuesto funciona”.
Si los ensayos clínicos de más envergadura confirman la eficacia del IONIS-HTTRx, estaremos ante un gran avance médico, ya que, hasta ahora, los tratamientos conocidos combatían los síntomas de esta dolencia, pero no la frenaban. Es cierto que el compuesto no la cura, pero sí podría evitar el agravamiento de sus síntomas más devastadores.
¿Qué es la enfermedad de Huntington?
Las personas que sufren esta dolencia neurodegenerativa nacen con la alteración genética que la desencadena, pero los síntomas no suelen aparecer hasta pasados de 30 a 40 años. Los primeros indicios incluyen problemas de equilibrio, torpeza y movimientos descontrolados. El avance del mal puede llegar a impedir que el afectado camine, hable o trague. Algunos enfermos dejan de reconocer a sus familiares, de forma similar a la de quienes padecen demencia.
Los afectados no suelen sobrevivir más de 20 años tras el diagnóstico de esta patología neurodegenerativa hereditaria, incurable e incapacitante que reúne síntomas que la asemejan al alzhéimer, el párkinson y la esclerosis lateral amiotrófica (ELA).
Image copyrightJAMES GALLAGHERImage captionPeter has Huntington's disease and his siblings Sandy and Frank also have the gene
The defect that causes the neurodegenerative disease Huntington's has been corrected in patients for the first time, the BBC has learned.
An experimental drug, injected into spinal fluid, safely lowered levels of toxic proteins in the brain.
The research team, at University College London, say there is now hope the deadly disease can be stopped.
Experts say it could be the biggest breakthrough in neurodegenerative diseases for 50 years.
Huntington's is one of the most devastating diseases.
Some patients described it as Parkinson's, Alzheimer's and motor neurone disease rolled into one.
Peter Allen, 51, is in the early stages of Huntington's and took part in the trial: "You end up in almost a vegetative state, it's a horrible end."
Huntington's blights families. Peter has seen his mum Stephanie, uncle Keith and grandmother Olive die from it.
Tests show his sister Sandy and brother Frank will develop the disease.
The three siblings have eight children - all young adults, each of whom has a 50-50 chance of developing the disease.
Worse-and-worse
The unstoppable death of brain cells in Huntington's leaves patients in permanent decline, affecting their movement, behaviour, memory and ability to think clearly.
Peter, from Essex, told me: "It's so difficult to have that degenerative thing in you.
"You know the last day was better than the next one's going to be."
Huntington's generally affects people in their prime - in their 30s and 40s
Patients die around 10 to 20 years after symptoms start
About 8,500 people in the UK have Huntington's and a further 25,000 will develop it when they are older
Huntington's is caused by an error in a section of DNA called the huntingtin gene.
Normally this contains the instructions for making a protein, called huntingtin, which is vital for brain development.
But a genetic error corrupts the protein and turns it into a killer of brain cells.
The treatment is designed to silence the gene.
On the trial, 46 patients had the drug injected into the fluid that bathes the brain and spinal cord.
Doctors did not know what would happen. One fear was the injections could have caused fatal meningitis.
But the first in-human trial showed the drug was safe, well tolerated by patients and crucially reduced the levels of huntingtin in the brain.
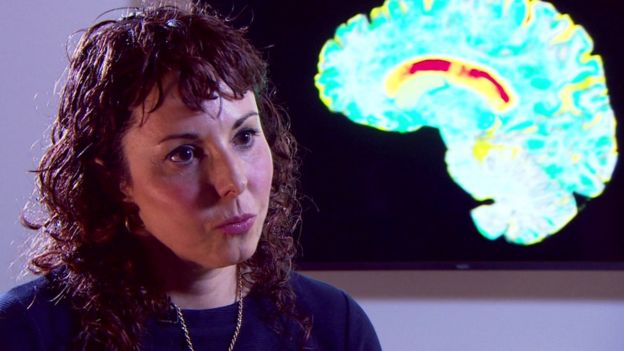
Image captionProf Sarah Tabrizi , from the UCL Institute of Neurology, led the trials.
Prof Sarah Tabrizi, the lead researcher and director of the Huntington's Disease Centre at UCL, told the BBC: "I've been seeing patients in clinic for nearly 20 years, I've seen many of my patients over that time die.
"For the first time we have the potential, we have the hope, of a therapy that one day may slow or prevent Huntington's disease.
"This is of groundbreaking importance for patients and families."
Doctors are not calling this a cure. They still need vital long-term data to show whether lowering levels of huntingtin will change the course of the disease.
The animal research suggests it would. Some motor function even recovered in those experiments.
Image copyrightJAMES GALLAGHERImage captionSandy Sterne, Peter Allen, Hayley Allen, Frank Allen, Annie Allen and Dermot Sterne
Peter, Sandy and Frank - as well as their partners Annie, Dermot and Hayley - have always promised their children they will not need to worry about Huntington's as there will be a treatment in time for them.
Peter told the BBC: "I'm the luckiest person in the world to be sitting here on the verge of having that.
"Hopefully that will be made available to everybody, to my brothers and sisters and fundamentally my children."
He, along with the other trial participants, can continue taking the drug as part of the next wave of trials.
They will set out to show whether the disease can be slowed, and ultimately prevented, by treating Huntington's disease carriers before they develop any symptoms.
Prof John Hardy, who was awarded the Breakthrough Prize for his work on Alzheimer's, told the BBC: "I really think this is, potentially, the biggest breakthrough in neurodegenerative disease in the past 50 years.
"That sounds like hyperbole - in a year I might be embarrassed by saying that - but that's how I feel at the moment."
The UCL scientist, who was not involved in the research, says the same approach might be possible in other neurodegenerative diseases that feature the build-up of toxic proteins in the brain.
The protein synuclein is implicated in Parkinson's while amyloid and tau seem to have a role in dementias.
Off the back of this research, trials are planned using gene-silencing to lower the levels of tau.
Prof Giovanna Mallucci, who discovered the first chemical to prevent the death of brain tissue in any neurodegenerative disease, said the trial was a "tremendous step forward" for patients and there was now "real room for optimism".
But Prof Mallucci, who is the associate director of UK Dementia Research Institute at the University of Cambridge, cautioned it was still a big leap to expect gene-silencing to work in other neurodegenerative diseases.
She told the BBC: "The case for these is not as clear-cut as for Huntington's disease, they are more complex and less well understood.
"But the principle that a gene, any gene affecting disease progression and susceptibility, can be safely modified in this way in humans is very exciting and builds momentum and confidence in pursuing these avenues for potential treatments."
The full details of the trial will be presented to scientists and published next year.
The therapy was developed by Ionis Pharmaceuticals, which said the drug had "substantially exceeded" expectations, and the licence has now been sold to Roche.
En un anuncio que puede que sea uno de los mayores avances en la enfermedad de Huntington, desde el descubrimiento del gen de la EH en 1993, las compañías Ionis y Roche anunciaron hoy que el primer estudio con humanos de un fármacos que reduce la huntingtina, Ionis-HTTRx, demuestra que reduce la huntingtina mutada en el sistema nervioso y que es segura y bien tolerada.
¿De qué se trata todo esto de bajar la huntingtina?
La terapia que más nos entusiasma para la enfermedad de Huntington se llamareducción de la huntingtina. También puede que usted lo conozca comosilenciamiento del gen, pero la disminución de la huntingtina es más precisa, como explicaremos.
Ionis pharmaceuticals ha dado a Roche la licencia de IONIS-HTTRx tras este exitoso estudio en Fase 1/2a
Todos tenemos dos copias del gen de la EH, uno heredado de la madre y el otro del padre. En las personas que desarrollarán EH, una de estas copias del gen de la EH se modifica, omutade una manera muy específica.
Justo cerca del comienzo del gen de la EH se encuentra una secuencia repetitiva que se lee, en el código utilizado por los científicos para describir el ADN,C-A-G. Las personas que no desarrollan la enfermedad de Huntington tienen alrededor de 20 repeticiones de esta secuencia, mientras que en las personas que desarrollarán la EH, es más larga, normalmente 40repeticiones CAGo más.
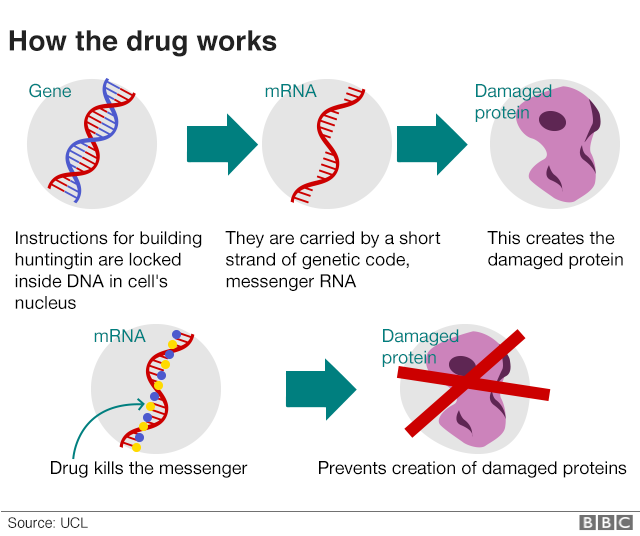
Nuestras células usan genes como recetas para construir proteínas: pequeñas máquinas moleculares que hacen cosas útiles en las células. Cuando una célula necesita producir más cantidad de una cierta proteína, se hacen copias de las instrucciones en una sustancia química estrechamente relacionada con el ADN, llamadaARN. Los científicos llaman a esta copia de un gen elARN mensajeroporque transporta la información de cada gen desde el ADN a las máquinas de construcción de proteínas de las células.
Esto significa que hay más de un lugar en la célula donde podemos encontrar la información de la mutación de la EH: la repetición anormalmente larga que se encuentra en el ADN de las personas también se copia en elARN mensajero. En última instancia, las células usan este mensaje delARNcomo instrucciones para construir una proteína: laproteína huntingtina.
La mayoría de los estudios sobre la EH sugieren que es laproteína huntingtina, no el gen o el mensajero, la que hace que las células cerebrales funcionen mal y mueran en personas con EH. Pero lo que sabemos con certeza es que cada persona con EH tiene una copia mutada del gen EH, que actúa como el anteproyecto de la proteína tóxica. Esto hace que el gen mutado de la EH sea el enemigo número uno para aquellos de nosotros que trabajamos para desarrollar nuevas terapias.
El rápido avance de la ciencia en las últimas décadas ha proporcionado a los científicos una gran caja de herramientas para cerrar selectivamente genes específicos. Algunas técnicas, como losoligonucleótidos antisentido, han existido durante décadas. Las técnicas más nuevas, especialmente las herramientas de edición genómica comoCRISPR/Cas9, sólo se han descubierto y desarrollado en los últimos años.
Si bien los detalles de las tecnologías difieren, en el mundo EH, todos tienen una potencial aplicación interesante: reducir la cantidad deproteína huntingtina. En numerosas pruebas en animales, usando una amplia gama de estas herramientas de silenciamiento, cuando los investigadores desactivaron el gen de la huntingtina anormal, los modelos animales de EH mejoraron, o nunca llegaron a enfermar.
Es genial, pero a nadie le importa curar la enfermedad de Huntington en un ratón, una mosca de la fruta o un gusano. Queremos curar la EH en la especie que más nos importa: los seres humanos con EH.
Recordatorio: ¿cuál es la historia de este fármaco y delensayo clínico?
«En el estudio de Fase 1/2a, se observaron reducciones dependientes de dosis de huntingtina mutada entre pacientes tratados con IONIS-HTTRx»
Entre todas las tecnologías de reducción de huntingtina que existen, el enfoque más desarrollado se denominaoligonucleótidos antisentidooASO. Se trata de piezas de ADN cortas, personalizadas y químicamente modificadas que pueden entrar libremente en las células. Una vez dentro, localizan y ayudan a destruir unARN mensajeroespecífico, en este caso, el que indica a las células sobre cómo fabricar laproteína huntingtina.
Ionis Pharmaceuticals, en Carlsbad, California, ha estado desarrollandoASOspara una variedad de enfermedades durante décadas. Hace años, se dieron cuenta de que la EH era perfecta para su tecnología, porque sabemos que si, en animales, reducimos los niveles de laproteína huntingtinaen el cerebro, mejoramos sus síntomas similares a la EH.
El año pasado Ionis tuvo un éxito masivo con un ASO para otra enfermedad cerebral llamadaatrofia muscular espinal(SMA). Este estudio quería comprobar si un ASO administrado a través líquido cefalorraquídeo podría ayudar a mejorar la enfermedad de los bebés que nacen con esta horrible enfermedad mortal. La misma tecnología básica, pero apuntando a un gen diferente.
A los niños con SMA les fue tan bien en el estudio de Ionis que tuvieron quedetener el estudio antes de tiempo, para que todos los niños del estudio, incluidos los que recibíanplacebo, pudieran recibir el medicamento. Básicamente, si la enfermedad hubiera tomado su curso normal, los niños se habrían debilitado progresivamente y hubieran muerto. Pero muchos de los niños tratados con el medicamento se volvían más fuertes y vivían mucho más tiempo.
El medicamento SMA de Ionis se aprobó posteriormente en los EE. UU., la UE y en muchos otros países, y ahora se está administrando a niños con SMA en todo el mundo.
Entonces, ¿qué hay de la EH?
Ionis ha estado trabajando en unoligonucleótido antisentido(ASO) para la EH desde comienzos de la década de 2000, primero en células simples y luego se ha mudado a varias especies de animales diferentes. Los efectos que estaban viendo eran prometedores y las pruebas en humanos se convirtieron en una posibilidad real. En 2013 cuando el gigante farmacéutico Roche anunció una asociación con Ionis para desarrollar el medicamento ASO para la EH, que ellos llamanIONIS-HTTRx, se adelantó el estudio. Esto trajo enormes recursos y experiencia en Roche para enfrentar el problema de la enfermedad de Huntington.
En julio de 2015, comenzó el estudio farmacéutico más emocionante hasta el momento en la enfermedad de Huntington, en la que un ASO diseñado para reducir la producción deproteína huntingtinarealmente se administraba a personas con ED. El ensayo se diseñó para evaluar la seguridad del fármaco y determinar si el fármaco podría hacer para lo que estaba diseñado: reducir la producción de laproteína huntingtina. Estábamos muy entusiasmados con el inicio de este ensayo y escribimos sobre el lanzamiento del ensayoaquí.
En cualquier esfuerzo de desarrollo de medicamentos, el primer objetivo debe ser garantizar que el medicamento no tenga efectos secundarios tóxicos. La historia nos proporciona muchos ejemplos de medicamentos que parecían una buena idea, pero que tenían efectos secundarios inesperados cuando se administraban a personas.
Se observaron reducciones dependientes de dosis de la proteína huntingtina mutada en el líquido cefalorraquídeo de pacientes que recibieron el medicamento
Con esto en mente, Ionis y Roche diseñaron un estudio cuyo objetivo principal era determinar si el medicamento es seguro cuando se administra a personas, lo que tiene que ser el primer paso en el proceso de desarrollo de fármacos.
Este primer estudio incluyó a 46 personas con síntomas iniciales de la EH en Alemania, Canadá y el Reino Unido. La prueba comenzó en julio de 2015 y estaba previsto que finalizara en noviembre de 2017. Como pueden ver, se cumplieron los plazos, lo que no siempre ocurre.
Antes de hablar sobre los resultados, hay algunos detalles importantes que las personas deben tener en cuenta. Primero, los medicamentosASOsno ingresan al cerebro si se ingieren en forma de píldora. Como consecuencia, los medicamentos ASO para enfermedades cerebrales se administran inyectándolos en la base de la columna vertebral, utilizando una técnica llamadapunción lumbar. Suena un poco aterrador, pero en realidad es un procedimiento muy habitual, realizado miles de veces al día en hospitales de todo el mundo.
Segundo, este estudio incluyó un brazoplacebo. Eso significa que algunos de los participantes realizaron todos los pasos, pero recibieron inyecciones sin medicamento. Este es un componente absolutamente crítico de los ensayos: si no tenemos un grupo de personas sin medicamento, ¿cómo podemos estar seguros de que los cambios que observamos se deben al fármaco y no a algún otro factor?
Por último, dosis. Cada vez que los investigadores administran un medicamento a las personas por primera vez, comienzan con una dosis muy baja. En un ensayo como este, formalmente llamado estudio dedosis ascendente múltiple, los primeros participantes reciben una dosis baja y luego los participantes que se unen posteriormente reciben dosis más altas del medicamento. Esto permite a los médicos monitorizar cuidadosamente a las personas con cada nueva dosis, por lo que los efectos negativos del tratamiento se identifican pronto.
¿Qué ha ocurrido ahora?
El lunes, 11 de diciembre, Ionis publicó un comunicado de prensa describiendo los principales resultados del primer estudio de IONIS-HTTRx. El titular fue: “Ionis pharmaceuticals facilita la licencia de IONIS-HTTRx a su socio tras el EXITOSO estudio de Fase 1/2a en pacientes con la enfermedad de Huntington”. También dijo: “Se han observado disminuciones dependientes de la dosis deproteína huntingtinamutada”.
Si usted se pregunta si esto es emocionante, les diremos que los dos editores de HDBuzz se deleitaron con un poco de baile feliz cuando vieron el comunicado de prensa. ¡Esrealmenteuna gran noticia!
Pronto explicaremos por qué esto es tan emocionante, pero hay algunas cosas que se deben tener en cuenta.
«Ahora la clave es pasar rápidamente a un estudio más grande para probar si IONIS-HTTRx retrasa la progresión de la enfermedad»
Primero - seguridad. Ionis y Roche monitorizaron muy cuidadosamente a los pacientes incluidos en el estudio para buscar signos de que el medicamento no es seguro. En el comunicado de prensa, Ionis informa: “el perfil de seguridad y tolerabilidad de IONIS-HTTRx observado en el estudio en Fase 1/2aapoya que se pueda continuar desarrollando”. Eso significa que no se observaron problemas de seguridad significativos en los participantes, por lo que el primer obstáculo para este medicamento en la EH se ha eliminado y podemos pasar a los siguientes pasos.
Recuerde: este ensayo no fue diseñado para demostrar que IONIS-HTTRx ayuda con los síntomas de la EH o la progresión. El objetivo principal de este estudio fue establecer que el medicamento esseguro. La primera vez que se pone un nuevo medicamento en el cuerpo de alguien, quiere exponer a la menor cantidad posible de personas, en caso de que surjan problemas de seguridad inesperados.
Además, recuerde que este estudio fue breve: cada paciente sólo recibió 4 meses de inyecciones. Este es un tiempo demasiado corto para buscar cambios en la tasa de progresión de la EH. Incluso si IONIS-HTTRx resulta ser un fármaco maravilloso, el impacto en los síntomas después de sólo 4 meses de tratamiento podría ser mínimo, y no esperaríamos detectarlos en un estudio tan pequeño.
Entonces, y este es un mensaje realmente importante,no sabemos aún si el fármaco mejoró los síntomas de la EH en personas.
Sin embargo, el estudio fue capaz de ir más allá de la seguridad de una manera importante. Cada vez que los voluntarios en el ensayo recibieron una dosis del medicamento, se tomó una muestra de su líquido cefalorraquídeo, que baña el cerebro y la médula espinal.
El trabajo previo había demostrado que los niveles de laproteína huntingtinase pueden medir en el líquido cefalorraquídeo. Parece que, como las células se enferman durante el curso de la EH, parte de su contenido se vierte en este líquido, que circula por el cerebro.
Dado que el objetivo de lasterapiasreductoras de la huntingtina como IONIS-HTTRx es reducir la cantidad deproteína huntingtinaen las células cerebrales vulnerables, en teoría, esto nos da una excelente manera de saber si el medicamento está haciendo lo que debe hacer. Simplemente medimos los niveles deproteína huntingtinaen el líquido cefalorraquídeo antes y después del tratamiento con el medicamento.
Creemos que esta es la noticia más emocionante del comunicado de prensa de hoy de Ionis.
Apaga el editor del genoma cuando hayas terminado
La edición del genoma CRISPR acaba de recibir un apagado. Cortamos el bombo para explorar la tecnología en EH
Laedición del genomaes una frontera muy discutida en la ciencia médica en este momento, teniendo en cuenta que la ‘cirugía del ADN’ tiene el potencial de tratar o curar enfermedades genéticas como la de Huntington. Aquí vamos a ver lo que esta tecnología puede hacer actualmente y discutimos los desafíos que todavía se encuentran en el camino. También discutiremos cómo un equipo de científicos suizos ha desarrollado recientemente una forma de desconectar la maquinaria deedición del genomauna vez que ha hecho su trabajo.
Primero algunos conceptos básicos
Todos estamos compuestos de células y cada célula contiene una copia completa de nuestro ADN. Nuestro ADN es el manual de instrucciones para nuestros cuerpos. Está formado por cuatro “letras” químicas: A, T, G y C. El manual de instrucciones completo se conoce comogenoma. Nuestras células leen la secuencia de letras químicas en el ADN para producir proteínas, y el ADN correspondiente a una proteína se llamagen.
La edición del genoma utiliza máquinas de proteínas para cortar el ADN en lugares precisos. Sin embargo, usarlo para editar genes en las células cerebrales es complicado y arriesgado. Y en realidad no usa brazos robóticos.
¿Qué es la edición degenoma?
La enfermedad de Huntington está causada por una mutación en el gen que es la receta de una proteína llamadahuntingtina. En personas con EH, la secuencia CAG se repite demasiadas veces al comienzo del gen. Eso hace que las células fabriquen una proteína dañina -huntingtina mutada.
¿No sería increíble si pudiéramos cambiar ese trozo de ADN a la normalidad? Esta idea no es nueva, pero recientemente se han desarrollado herramientas que algún día podrían permitir la edición de ADN en personas.
Laedición del genomautiliza proteínas llamadasnucleasas, que son máquinas moleculares que cortan el ADN. La tecnología que ha sido noticia recientemente esCRISPR. Su historia se remonta a principios de la década de los 1990 cuando los investigadores encontraron grupos extraños de letras de ADN repetidas en las bacterias. Los llamaronCRISPR, pero no sabían en ese momento lo que hacían. Un poco más tarde, en 2002, los científicos detectaron que hay instrucciones de ADN para hacer una nucleasa muy cerca de estas repeticiones. Llamamos a estas nucleasas ‘Cas’. Luego, en 2005, otra pieza del rompecabezas encontró su lugar cuando los investigadores descubrieron que las secuencias cortas entre las repeticiones no provenían de las propias bacterias, sino que en realidad eran ADN virales que se habían agregado algenomabacteriano después de una infección.
Resulta que la combinación deCRISPRyCas(CRISPR/Cas) es en realidad un sistema inmune bacteriano, un arma que utilizan contra los virus. Cuando un virus infecta una célula bacteriana, la bacteria roba un poco de su ADN y lo inserta en su propiogenomaentre las repeticiones deCRISPR. Toda la secuencia (CRISPR, ADN viral y máquina de corte por nucleasa) se convierte en un arma que puede reconocer el ADN del virus invasor y cortarlo, previniendo la infección.
Finalmente, en 2012, Jennifer Doudna y Emmanuelle Charpentier demostraron que ajustando la secuencia de ADN puedes hacer queCascorte el ADN en el punto que desees. Esa parte es como el sistema de focalización de laCasnucleasa - ¡listo! ¡tenemos una herramienta deedición del genomahecha a medida!
Convertir laedición del genomaen un tratamiento
Las células humanas no tienenCRISPRoCas, por lo que para editar elgenomahumano, primero tienes que enseñar a las células cómo hacer estas herramientas de edición de genes. Para hacer eso, los científicos meten la receta de ADN para hacerCRISPRyCasen un virus inocuo e infectan las células con él. El virus inyecta el ADN en las células. Las células fabrican las herramientas de ediciónCRISPRyCas, que luego se incorporan alpropioADN de la célula, cortándola en la ubicación deseada.
Un gran desafío es asegurarse de queCasno alcance el objetivo equivocado. Si hay una secuencia de ADN en otro lugar que sea muy similar,Caspodría cortar eso también. Esto significa que, en el proceso de tratar de corregir una mutación en un gen, podría introducir otra en otro lugar, y eso podría causar una enfermedad completamente nueva.
«El sistema de edición del genoma KamiCas9 primero desactiva el gen de la huntingtina, y luego, unas cuatro semanas después, se apaga»
Edición del genomapara tratar enfermedades
Laedición del genomatiene el potencial de curar muchas enfermedades. La investigación se encuentra en una etapa inicial, particularmente en humanos. En un estudio reciente, los investigadores chinos utilizaronCRISPR/Casen embriones humanos para corregir una mutación que causa la enfermedad de la sangre beta-talasemia. Los embriones no fueron implantados, pero demostraron que elgenomahumano puede ser editado.
Usar laedición del genomapara tratar la enfermedad de Huntington
Actualmente se está llevando a cabo un emocionante estudio de “disminución de huntingtina” utilizando un fármaco llamado oligonucleótido antisentido (ASO) para reducir la cantidad deproteína huntingtinaen las células cerebrales. Este método a veces se denomina “silenciamiento del gen”, pero estonoesedición del genoma, porque el medicamento no altera el ADN del cerebro.
Laedición del genomairía un paso más allá al tratar la enfermedad de Huntington a nivel del ADN. Hay varias formas de abordar esto. Idealmente, sería posible recortar la repetición de CAG larga a una longitud normal. Sin embargo, aunqueCRISPR/Casactualmente es bueno para cambiar letras individuales en el ADN, todavía no puede enfocarse específicamente en el gen expandido y reducir el número de repeticiones de CAG. Un enfoque alternativo es introducir el equivalente genético de un STOP en el genHTT, por lo que el gen no produciría la proteína.
En teoría, laedición del genomadetendría de forma permanente y completa fabricación de la proteína. Esto puede sonar bien, pero es potencialmente un arma de doble filo, porque una vez hecho no puede revertirse, por lo que si algo sale mal, podría tener efectos duraderos.
RefinandoCRISPR/Casen la enfermedad de Huntington
Una vez que el ADN deCRISPRyCasse ha insertado en ungenoma, permanece allí para siempre. Esto significa que las células continuarán produciendoCasnucleasas, aunque solo sea necesario para hacer un trabajo: cortar el ADN de la célula en la que está flotando. Después de eso, ya no es necesario.
Tarde o temprano, existe el riesgo de que laCasnucleasa corte el ADN en alguna parte que no debería, introduciendo una mutación que podría causar una enfermedad. Además, recuerda queCasoriginalmente vino de una bacteria. Esto significa que el sistema inmune humano podría reconocerlo como invasor y tratar de atacarlo, produciendo una reacción inmune peligrosa.
Un riesgo de la edición de genes es que podría impactar accidentalmente al objetivo equivocado, alterando los genes que no queremos cambiar
Idealmente, queremos un tratamientoCRISPR/Casque solo funcione muy brevemente, editando el ADN de la manera elegida y luego lo apaguemos.
El equipo de Nicole Déglon de la Universidad de Lausana en Suiza ha desarrollado una forma de conseguir que haga precisamente eso. Desarrollaron una forma de desactivarCasuna vez que terminó de editar el genHTT, lo que reduce las posibilidades de que desencadene una reacción inmune o corte en algún lugar que no debería.
La genial idea del equipo suizo fue hacer una máquinaCRISPR/Casque se dirija al gen de la huntingtina, pero con una secuenciaextradeCRISPRque también hace que laCasnucleasa se dirija a su propio ADN. Cuando corta su propio ADN, el sistema se inactiva por sí mismo.
Esta secuenciaCRISPRadicional, a la que denominaron “KamiCas9” (sí, el nombre es un juego de palabras bastante dudoso sobre la palabra “kamikaze”), se produce a un ritmo mucho más lento que el de la huntingtina, por lo que su efecto se retrasa. Eso significa que primero desactiva el gen de la huntingtina, y luego, aproximadamente cuatro semanas después, el sistema deedición del genomase apaga. Las ediciones hechas al gen huntingtina durante las primeras cuatro semanas permanecerán para siempre, pero la desactivación eventual de laCasnucleasa reduce las posibilidades de efectos nocivos más adelante.
¿Dónde deja esto laedición del genoma?
Laedición del genomatiene un gran potencial para tratar una amplia gama de enfermedades. Sin embargo, si no se hace correctamente, también podría introducir problemas genéticos en el ADN humano que tendrían efectos incalculables para los pacientes y las generaciones futuras.
El equipo de Déglon ha hecho un avance importante al apagar el hardware de edición una vez que ha hecho su trabajo. Sin embargo, conseguir que el sistema de edición de genes en el cerebro humano siga siendo un gran desafío, y también lo es el riesgo de que se corte en el lugar equivocado antes de que se desactive.
Laedición del genomaes una tecnología emocionante que en el futuro podría ser una ruta para prevenir la enfermedad de Huntington, o incluso eliminar el riesgo para las generaciones futuras. Este nuevo interruptor de apagado es un ejemplo de cómo los científicos trabajan arduamente para mejorar las técnicas todo el tiempo. ¡El trabajo para preparar laedición del genomapara ayudar a las familias afectadas por la enfermedad de Huntington continúa!
El co-fundador de HDBuzz, Ed Wild, es un investigador del programa Ionis HTTRx y miembro de los Consejos Asesores Científicos de Ionis y Roche. Por eso este artículo fue escrito por Jeff Carroll. Jeff colabora con Ionis en estudios con ratones, pero no participó en este ensayo. Tamara Maiuri no tiene ningún conflicto de intereses para declarar.Más información sobre nuestra política de privacidad en las Preguntas frecuentes
Fundacion Verónica Ruiz
Enfermedades Neurodegenerativas y Enfermedad de Huntington México